Other Features¶
Data Formatting Help¶
Variables page of Metaboverse, launch the Format dataset module.Note
The statistical procedures used by this module assume data are normally distributed, such as is often the case with proteomics and metabolomics data. However, for transcriptomics data, which follow a negative binomial distribution, a package, such as DESeq2 or limma should be used. The resulting fold change and adjusted p-values should then be isolated from the results, exported into a tab-delimited file, and uploaded for use in Metaboverse. This formatting tool will display the distributions of each of the samples in the uploaded dataset to assist in verifying the distributions of the underlying data.
Warning
This Format Dataset tool from Metaboverse v0.10.1 onward will display the sample distributions within the uploaded datasets. Users should examine the samples to ensure their values follow normal distributions. If the data are not normally distributed, the user will need to utilize other statistical testing in preparing their dataset for Metaboverse.
Control or Experiment).Check Names button to cross-reference the names you provided with MetaboAnalyst to improve the chances that the metabolite correctly maps to the metabolic network.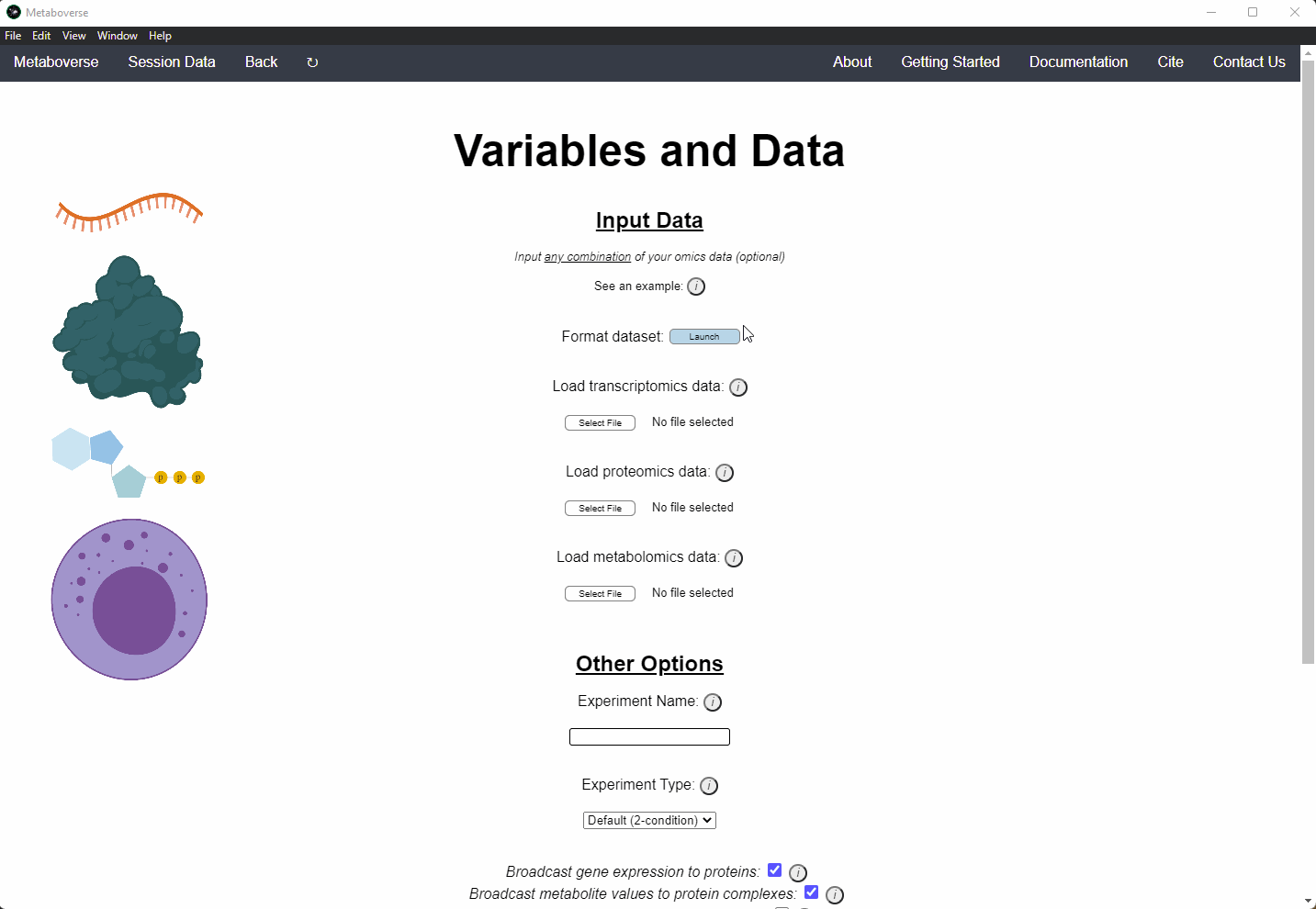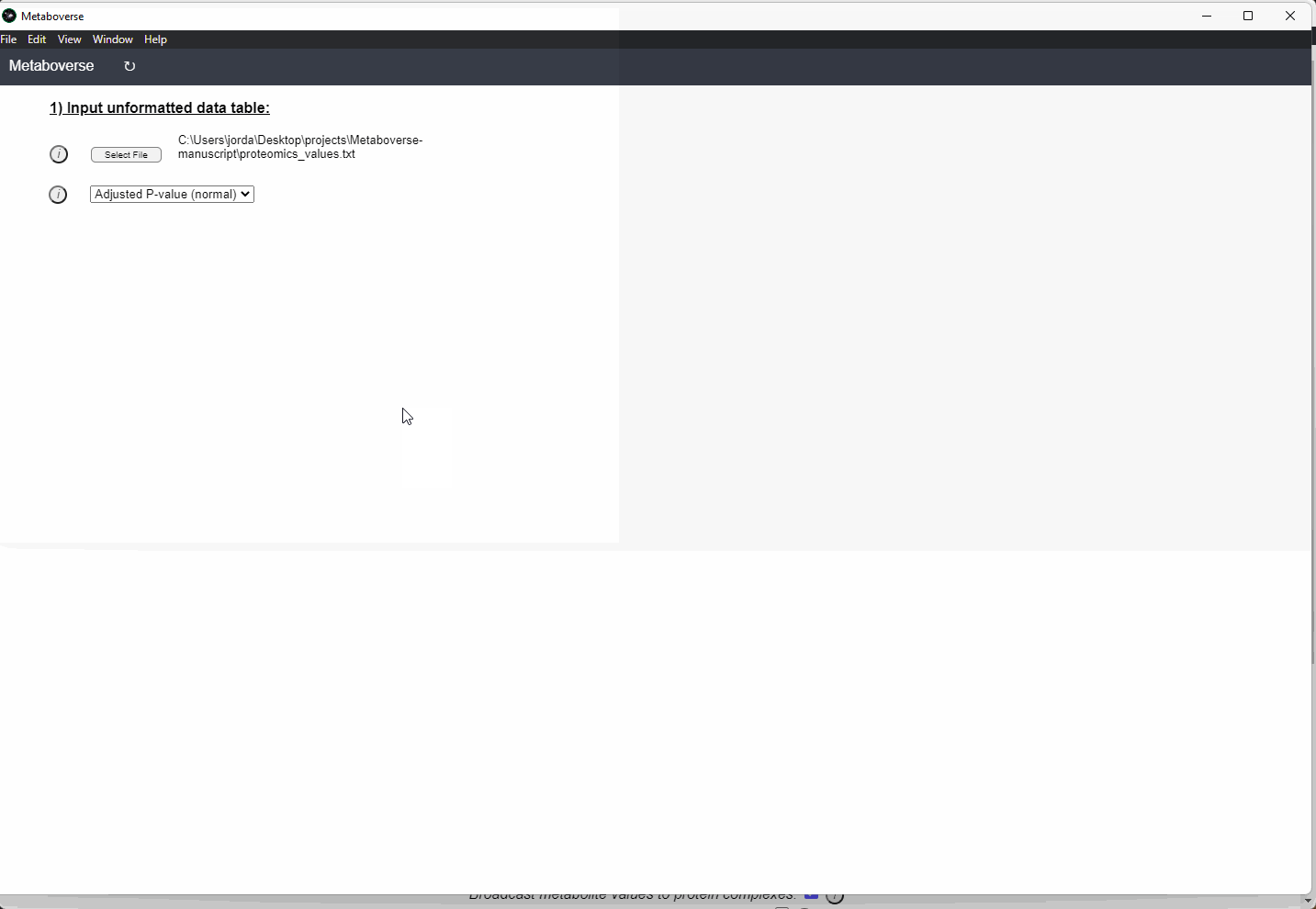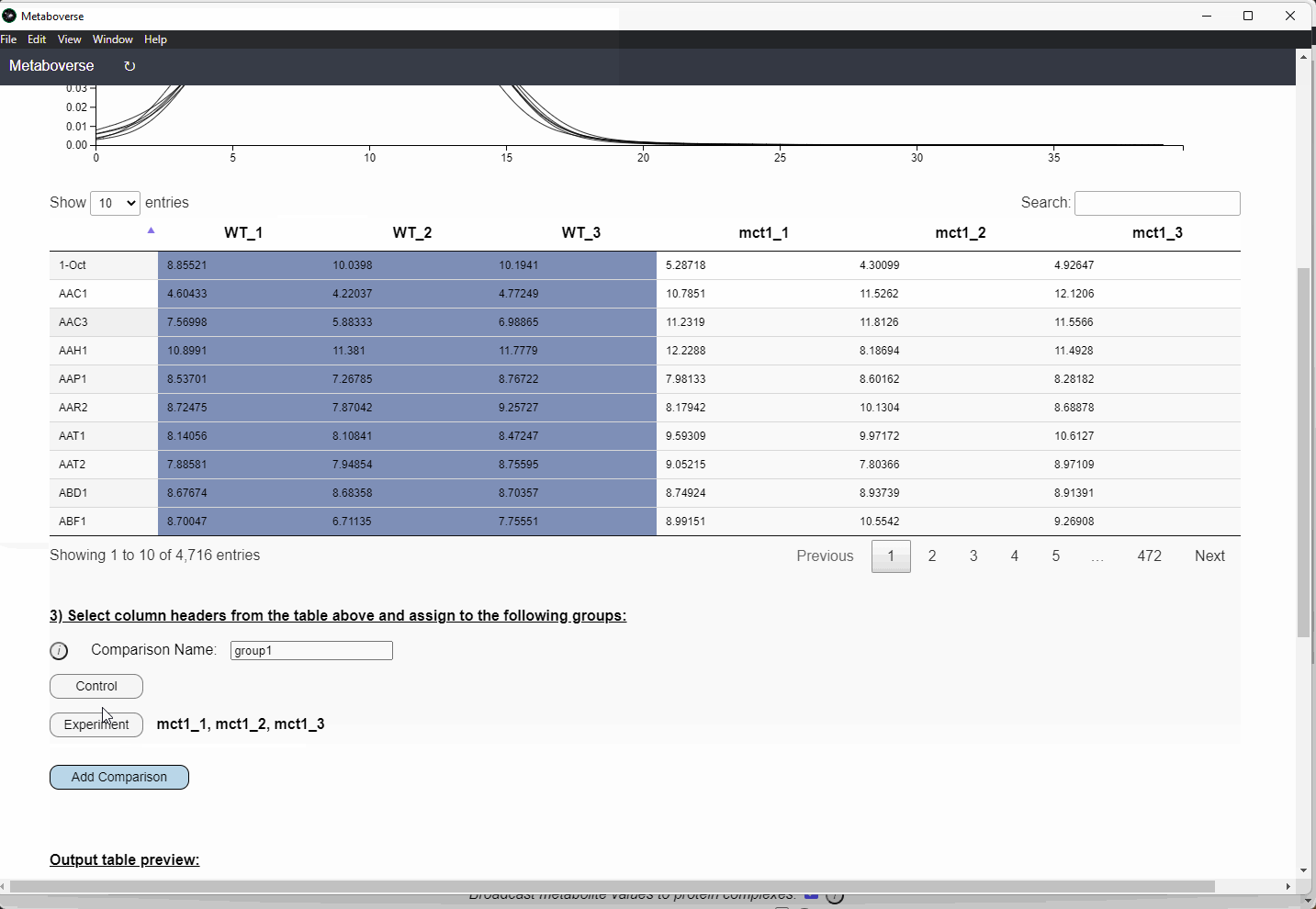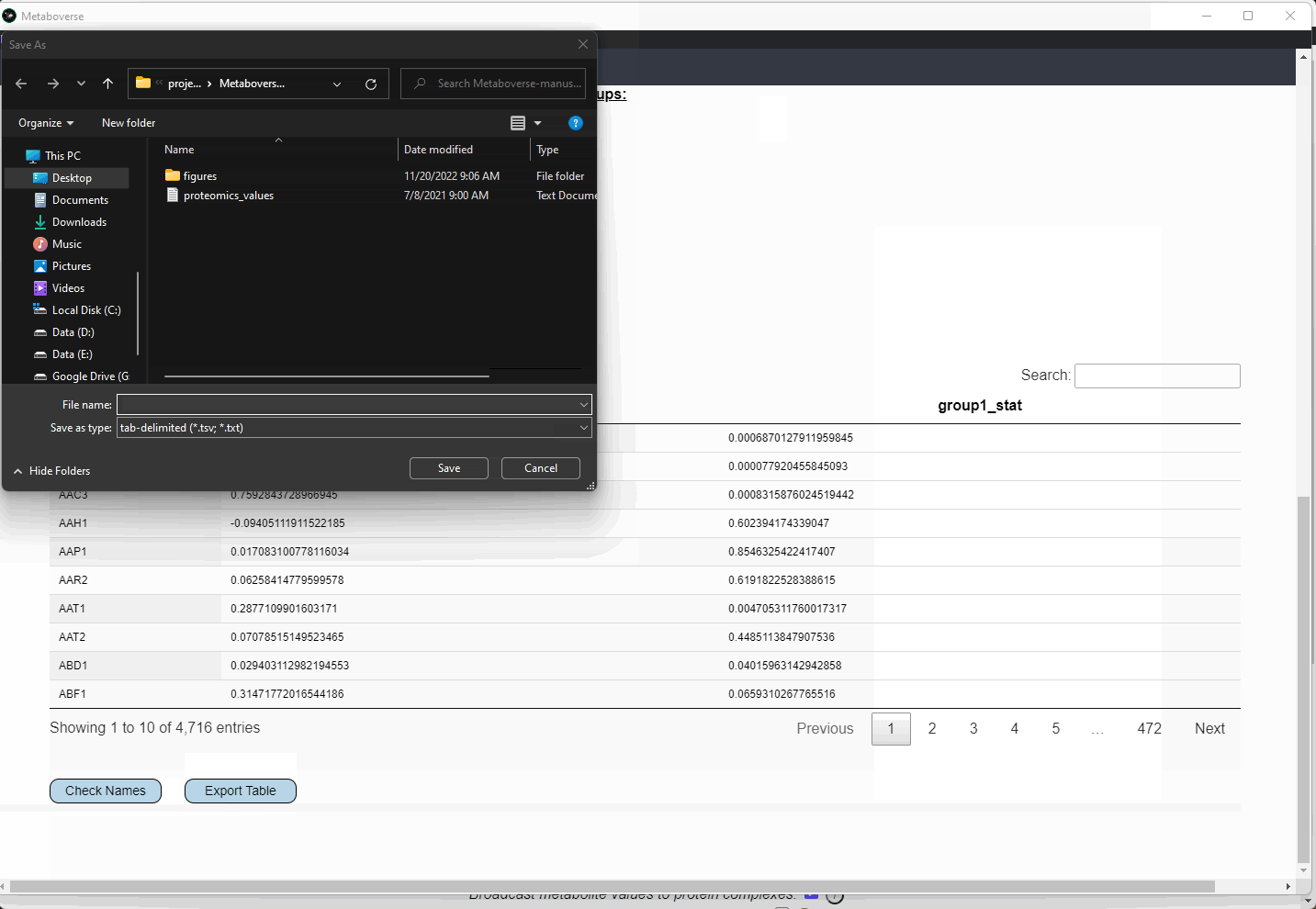
Tracing Metabolomics Data¶
ionization products with Escher-Trace and analyzing derived steady-state metabolomics data (i.e. M+0) with Metaboverse’s reaction pattern search engine. Cross-referencing the outputs of these two tools may then provide biological clues for their system, such as to the downstream outcomes of differential metabolite behavior.